
肺高压是以肺血管明显重构和肺血管负荷逐渐增加,进而导致右心室肥厚和重构为特征的综合征。如果肺高压不进行治疗,患者可因右心室衰竭死亡。肺高压目前的血流动力学定义为静息状态下平均肺动脉压>20 mmHg(通过右心导管测定)1。肺血管疾病引起的毛细血管前肺高压进一步定义为肺血管阻力增加≥3个Wood单位(WU),而单纯毛细血管后肺高压的肺血管阻力<3 WU,并且平均肺动脉压升高是由左心充盈压力升高所致的。
几种形式的肺高压在临床上分成5组(图1)。本文着重介绍相对罕见的肺动脉高压(第1组)。我们对肺动脉高压机制、自然史和遗传特征的理解,以及对其靶向治疗已取得了巨大进展。充分理解该综合征的病理生理学很重要,因为诊断该疾病需要通过全面临床检查来排除其他更常见的肺高压形式(对于后者,治疗基础疾病应是首要目标)。
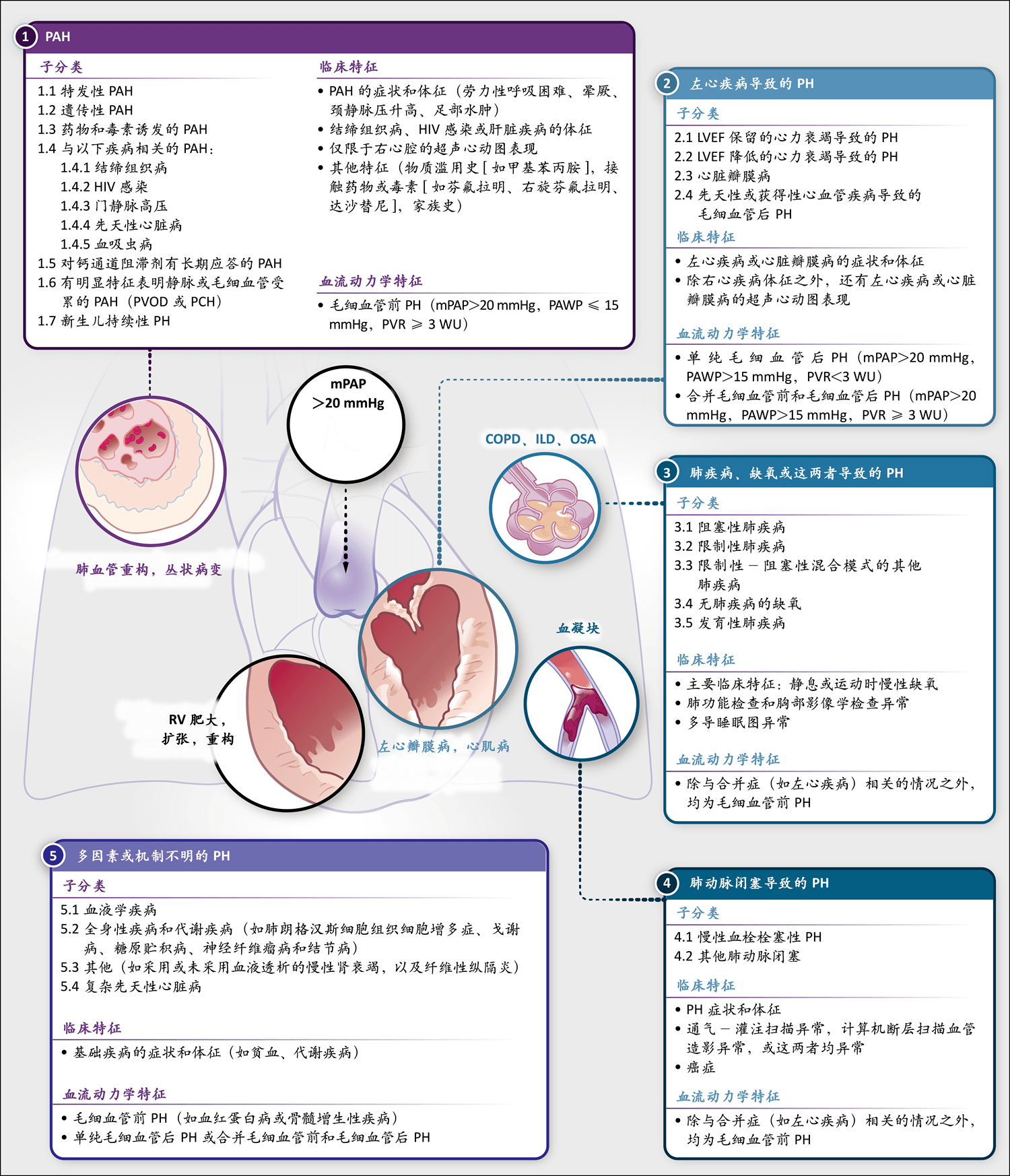
图中显示了基于世界肺高压研讨会(World Symposium on Pulmonary Hypertension)2018年会议的PH分类,以及各组疾病的临床特征和血流动力学特征1。COPD表示慢性阻塞性肺疾病,HIV表示人类免疫缺陷病毒,ILD表示间质性肺病,LVEF表示左心室射血分数,mPAP表示平均肺动脉压,OSA表示阻塞性睡眠呼吸暂停,PAH表示肺动脉高压,PAWP表示肺动脉楔压,PCH表示肺毛细血管瘤病,PVOD表示肺静脉闭塞性疾病,PVR表示肺血管阻力,RV表示右心室,WU表示Wood单位。
最早对肺高压的解剖学做出描述的是von Romberg2。Cournand和Richards在20世纪40年代对心脏和肺循环进行一系列生理学观察,是Forssmann在1929年首先于自己身上实施的人右心室导管插入术促使了他们的研究3。这三位研究人员因其开创性工作在1956年获得了诺贝尔生理学或医学奖。在其富有启发性诺贝尔奖领奖演讲中,Richards指出,作为他们共同工作的成果,“多种形式和程度的衰竭得到定义,衰竭对治疗的应答也得到评估4。”
1951年,Dresdale(Cournand和Richards的学生)及其同事首次发表了不明原因的肺高压(定义为“原发性肺动脉高压”)病例系列5。20世纪60年代,在集中出现与食欲抑制剂阿米雷司(aminorex)相关的原发性肺高压后,人们对这种疾病有了更深刻的认识6。这促使世界卫生组织于1973年第一次召开了肺高压专家会议,目的是规范原发性肺高压的临床和病理命名,这是首次尝试有组织的分类7。关于肺高压的第一次和第二次会议相隔25年,但此后每5年召开一次世界肺高压研讨会(World Symposium on Pulmonary Hypertension,WSPH)。会议进一步细化了肺高压的分类,根据相似的临床和病理检查结果及治疗应答情况将肺高压分成5组(关于历史背景的讨论见补充附录,补充附录与本文全文可在NEJM.org获取)。人们认识到在血流动力学和临床特征方面,肺动脉高压与直接影响肺动脉血管,并且已经有靶向治疗的其他疾病具有相似性,因此特发性肺动脉高压一词取代了原发性肺高压。
人们认识到部分人对这一疾病具有遗传易感性(家族性肺动脉高压),并在这一基础上里程碑性地发现了形态发生蛋白(BMP)受体2型编码基因(BMPR2)突变8,9。由于80%的家族性肺动脉高压病例和多达20%的散发病例有生殖细胞系BMPR2突变,并且由于已在各种基因中发现其他突变,因此家族性肺动脉高压一词更改为遗传性肺动脉高压。
在本世纪的前20年,由于人们对肺动脉高压的关注度和认识均增加,因此出现了一系列新的口服、注射和吸入药物。这些药物的研发是基于多项妥善实施的安慰剂对照研究(补充附录图S1)。
全球健康问题
肺高压患病率随WSPH分组不同而异。在西方国家,每100万人中有25人患肺动脉高压(第1组)(其中大多数是女性),每年发病率为每100万人中2~5例10。该疾病在老年男性中最为严重11,但在这一人群中不太常见,这一人群更易患第2组疾病。对于肺高压分类中的其他组,患病率因病因和疾病状态不同而异,但可能在世界各地都被大大低估12。
许多广泛存在的心肺系统疾病都并发肺高压,这大大增加了发病率和死亡率。由于先天性心脏病的发病率在世界各地(特别是在发展中国家)都很高13,因此估计全世界每100万人中有25例先天性心脏病引起的肺动脉高压14。心脏瓣膜病和左心疾病更为常见15,16,全世界可能有超过1亿人患左心疾病导致的肺高压(第2组)。
同样,慢性肺疾病(如慢性阻塞性肺疾病[全世界负担,>5亿例]和间质性肺病[估计发病率,10%~70%])可并发肺高压;肺高压患病率在晚期肺疾病患者中增加14。此外,超过1.4亿人生活在高海拔地区(高于2,500 m)17,但居住在高海拔地区或移居到这些地区的人中,慢性缺氧导致的肺高压患病率尚不明确。此外,高度流行的病毒感染(如人类免疫缺陷性病毒[HIV]感染)和寄生虫病(如血吸虫病),以及血红蛋白病(如镰状细胞病和地中海贫血)可并发肺高压。因此,非洲、亚洲、南美洲和中美洲的低收入和中等收入地区有大量这样的患者18,19。
因此,据估计,1%的世界人口和多达10%的65岁以上人口患肺高压14。此外,这些患者中有80%生活在发展中国家,而且由于治疗费用高昂20、缺乏获得批准的药物或者获得必需内科和外科支持的机会有限(如慢性血栓栓塞性肺动脉高压[第4组]的治疗),许多患者不太可能得到治疗12,14。
病理学特征
由于基础疾病的多样性,肺动脉高压的组织学特征复杂多变。但这一疾病具有共同的病理学特征,如肺远端血管三层重构(图2),包括内皮细胞、平滑肌细胞和成纤维细胞生长失控22,以及炎症细胞浸润23,主要累及直径为50~500 μm的毛细血管前血管。平滑肌细胞层也延伸到一般无肌层的末梢毛细血管。发生类似静脉重构的毛细血管后血管可能与特定的综合征相关,如肺静脉闭塞性疾病和肺毛细血管瘤病、硬皮病相关性肺动脉高压24、慢性血栓栓塞性肺高压25、血管重构可能始于毛细血管后隔室(postcapillary compartment)的第2组心脏病26。人们一直认为22累及小肌性动脉的原位血栓形成是由血小板激活和内皮完整性丧失引起的27。这些改变导致管腔狭窄或小血管完全闭塞。网状病变是肺动脉高压的共同特征,其原因可能是支气管动脉或穿过肺血管壁结构的营养血管发生吻合28(图2)。导致严重重构的事件尚未明确,但剪切应力、缺氧、自身免疫现象、病毒感染、药物和毒素或基因改变引起的内皮功能障碍可能会引发血管过度收缩、炎症和细胞生长失控27。人们观察到与肺动脉高压病变并列的高度组织化的淋巴样滤泡,T淋巴细胞和B淋巴细胞浸润29,以及与疾病严重程度相关的循环炎症标志物30,再加上自身免疫病或炎症性疾病常并发肺动脉高压,这些令我们相信炎症在肺动脉高压发病机制中发挥了作用23,31。
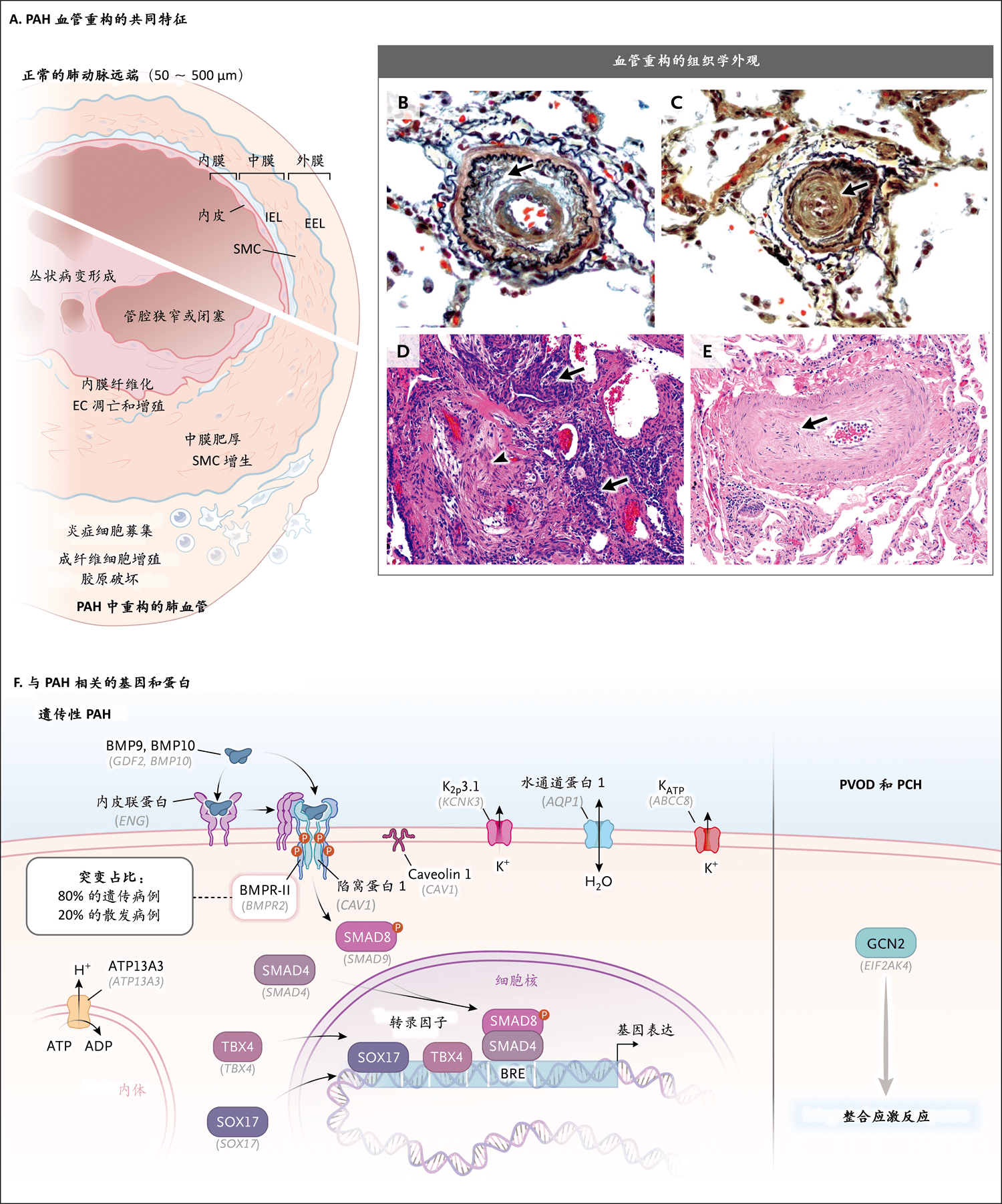
图A显示正常远端肺动脉和以血管壁三层(内膜、中膜和外膜[EEL表示外弹性层,IEL表示内弹性层,SMC表示平滑肌细胞])重构为特征的PAH血管。内皮细胞(EC)凋亡和增殖引起内膜增厚,平滑肌细胞增生引起内侧增厚,单核细胞、B淋巴细胞、T淋巴细胞和树突状细胞等炎症细胞浸润,以及胶原破坏促进外膜重构。在图B中,远端肺血管弹力纤维染色显示动脉腔明显狭窄,并且有同心圆样层状内膜增生和纤维化,以及IEL破坏(箭形)。图C中的弹力纤维染色显示肺小动脉完全闭塞(箭形)。图D(苏木精和伊红染色)显示丛状病变,近端为闭塞性病变(箭头),远端为过多且形态不良的毛细血管和大量内皮样细胞群(箭形)。图E(苏木精和伊红染色)显示PVOD特征性病变,伴内膜纤维化(箭形)和血管腔明显闭塞。图F(改编自Southgate等的论文21)显示转化生长因子β(TGF-β)超家族成员及遗传性PAH相关基因和蛋白的信号转导,包括膜结合受体,如骨形态发生蛋白受体Ⅱ型(BMPR2);激活蛋白A受体Ⅱ型样蛋白1(ACVRL1),又称激活蛋白受体样激酶1(ALK1);内皮联蛋白(ENG);陷窝蛋白-1(CAV1);以及转录因子SMAD4和SMAD8(SMAD4和SMAD9)。以细胞特异性方式介导骨形态发生蛋白(BMP)信号传导的是配体BMP9和BMP10与BMPR-Ⅱ异源复合物、同源1型受体(ACVRL1或ALK1)和内皮联蛋白(辅助受体,目前认为其可捕获配体,抑制受体结合,从而调节BMP的效应)之间的结合。受体复合物的激活导致受体和SMAD8的磷酸化,与SMAD4结合后转位到细胞核,调节含有BMP响应元件(BRE)的基因。KCNK3(钾通道亚家族K成员3[K2P3.1]编码基因)促进肺血管张力,并且是ATP敏感钾(KATP)通道(ABCC8)的亚基。SOX17和TBX4(器官发生的重要转录因子)也参与BMP信号传导。可能的阳离子转运ATP酶13A3(ATP13A3)与BMP信号传导无直接关系。然而,ATP13A3失活突变会破坏多胺稳态,导致内皮功能障碍,从而促进PAH发病。水通道蛋白1(AQP1,也不是TGF-β超家族成员)与β-联蛋白相互作用,激活与细胞生长相关的靶基因,进而介导肺动脉平滑肌细胞的迁移和增殖。GCN2(general control nonderepressible 2)(也称作EIF2AK4[真核细胞翻译起始因子2α激酶4])编码整合应激反应中的一个重要激酶,GCN2的双等位基因突变与PVOD和PCH的发病机制相关。
右心室
右心室功能是肺动脉高压患者临床结局和生存期的主要决定因素32。随着肺动脉高压进展,肺血管阻力增加至5~10倍,出现右心室肥厚、心腔扩张、脂肪沉积、纤维化和代谢性改变32。
右心室重构可能适应良好,表现为向心性肥厚、心肌微循环保留和心肌纤维化程度轻;也可能适应不良,表现为离心性肥厚、微血管稀少(导致氧供需失衡)和心肌纤维化32。对于导致上述变化或导致两种状态间转变的机制,目前知之甚少,但可能包括血管生成过程改变、从葡萄糖氧化转向糖酵解和脂肪酸氧化,以及线粒体生物能量学改变33。
作为评估右心室固有心肌功能和右心室-肺血管耦合的标准,带高保真电导导管的压力-容积环技术是一种有创检查,且需要专门的技术32。替代性无创技术(超声心动图或心脏磁共振成像[MRI])34-36仍需对照上述标准进行验证,但这些无创技术的确可预测结局34,37。最近在论文中被强调的方面包括右心室功能障碍的机制,目前缺乏右心室靶向疗法,以及研究工作正不断取得进展的知识缺口32,38。
在未患心血管疾病的人群中,女性的右心室射血分数优于男性39,这归因于性激素差异40和不同性别对某些药物(如磷酸二酯酶抑制剂和内皮素受体拮抗剂)的应答差异41。但仍需进一步研究。
对心肌细胞进行的体外研究帮助我们了解了心肌固有收缩能力,揭示了特发性或先天性心脏病相关肺动脉高压患者的收缩力亢进表型42,这与硬皮病相关性肺动脉高压患者的收缩力减退表型形成鲜明对比43。与右心室收缩力体内测定值相关的这些发现有可能解释后一组患者较差的临床结局和较短的生存期。这些表型的分子基础仍然缺乏研究。
遗传特征
2000年取得了重大突破,当时有两个独立的研究团队8,9发现了转化生长因子β(TGF-β)超家族成员BMPR2的杂合突变。这一突破结合基因技术的进步(例如全基因组和全外显子组测序),帮助我们进一步了解了某些基因在肺动脉高压发病机制中发挥的作用。
研究发现BMPR2突变见于约80%的家族性肺动脉高压患者(男性和女性患者的外显率不同)和多达20%的散发性肺动脉高压患者。之后很快又发现ACVRL1(编码激活蛋白A受体Ⅱ样蛋白1[也称为激活蛋白受体样激酶1])和ENG(编码内皮联蛋白)44,45突变见于遗传性出血性毛细血管扩张症(一种偶尔并发肺动脉高压的综合征)家族。ACVRL1和内皮联蛋白都通过二聚作用参与BMPR-Ⅱ信号传导(图2)。
对肺动脉高压患者大规模队列的进一步分析在BMPR2突变阴性的家族中发现了转录因子SMAD1、SMAD4和SMAD9编码基因46(复杂BMPR-Ⅱ下游信号传导的一部分)的其他突变21和其他基因的突变47,包括陷窝蛋白148(参与BMP受体的共定位)编码基因(CAV1)和钾通道亚家族K成员349(涉及膜电位维持和肺血管张力)编码基因(KCNK3)。TBX4(编码T-box转录因子4,是与小髌骨综合征相关的基因)突变见于一些有智力障碍和营养不良特征的儿童、他们父母中的一些人,以及另外一个肺动脉高压成人患者小队列50。在大队列中,BMPR2突变占多数(图S2)。其他新的突变涉及ATP13A3(编码ATP酶13A3)、SOX17(编码SRY-box 17,是先天性心脏病相关肺动脉高压的主要危险因素)51、AQP1(编码水通道蛋白1),以及GDF2(编码生长分化因子2,也称为BMP9)47。
EIF2AK4(编码真核细胞翻译起始因子2 α激酶4)双等位基因突变在遗传性肺毛细血管瘤病52和肺静脉闭塞性疾病病53中已有报道,而且见于多达25%的上述疾病散发病例。最近,生殖细胞系TET2(编码10-11易位[tet]甲基胞嘧啶双加氧酶2[DNA去甲基化的关键酶])突变在一个肺动脉高压患者大队列中有报道54。
BMPR-Ⅱ信号传导改变在肺动脉高压发病机制中的作用如何高估都不过分。目前发现的大多数突变均涉及BMPR2,或者涉及的基因是编码形成复合物或者与BMP或BMPR-Ⅱ信号传导相互作用的蛋白(图2)。BMPR-Ⅱ失活导致内皮功能障碍及增殖和凋亡之间的平衡改变,这是肺动脉高压的特征,这解释了人们为何日益关注旨在增加BMP9表达55或配体水平的疗法(如BMP9给药的临床前模型)56。
临床医师在伦理上有责任将遗传性疾病告知患者及其家人,尤其是特发性或遗传性肺动脉高压、肺静脉闭塞性疾病、肺毛细血管瘤病和先天性心脏病相关肺动脉高压。医师应考虑患者家人及其后代(他们可能是突变携带者)受到的影响,并由多学科专家团队提供筛查、遗传和心理咨询,并且为患者提供教育57。可应用数种负担得起的技术和平台同时检测多个基因。基因检测是数项国家和国际合作研究58及美国国立卫生研究院发起的PVDOMICS(肺血管病表型组学[Pulmonary Vascular disease Phenomics])项目59的任务,有望帮助我们进一步了解该疾病,并且通过特定靶点改进治疗。
诊断
病史和体格检查
肺动脉高压的症状不具有特异性(劳力性呼吸困难、疲劳、胸痛、液体潴留,以及晚期病例的晕厥),这是许多病例诊断延迟的原因。如果有基础疾病(如HIV感染,肝脏或结缔组织病,或者药物或毒素暴露史),则应该增加对肺动脉高压的怀疑(图1)。诊断该疾病的主要难度是排除其他类型的肺高压(对这些肺高压的治疗应集中于基础疾病),因此左心疾病或慢性肺疾病的危险因素或症状是很重要的考虑因素。
提示肺高压的体格检查结果包括肺动脉瓣第二音增强、三尖瓣反流杂音和右心室液体超负荷证据(如颈静脉压增高和足部水肿)。还有其他检查结果可能提示肺高压的潜在原因,包括慢性肝病或风湿性疾病的后遗症。
诊断检查
经胸超声心动图(TTE)是最重要的筛查项目(图3),它提供了一组衡量该疾病患病率、病因和严重程度的指标。这些指标包括右心腔扩张情况;是否有三尖瓣反流及其严重程度(可据此估算右心室收缩压);是否有心包积液;以及右心室容量和压力超负荷导致的异常间隔偏曲。TTE还可识别左心室收缩或舒张功能障碍及瓣膜异常,这些发现将焦点转移到第2组肺高压。除全血细胞计数和代谢检测组合之外,抗核抗体滴度测定和HIV血清学检测可能也有助于发现特定的基础疾病,如结缔组织病或HIV疾病。血清氨基末端利钠肽水平(作为非特异性心脏生物标志物)可纳入风险分层(见下面的讨论),因为其数值与肺动脉高压严重程度密切相关,可用于预测生存期61。
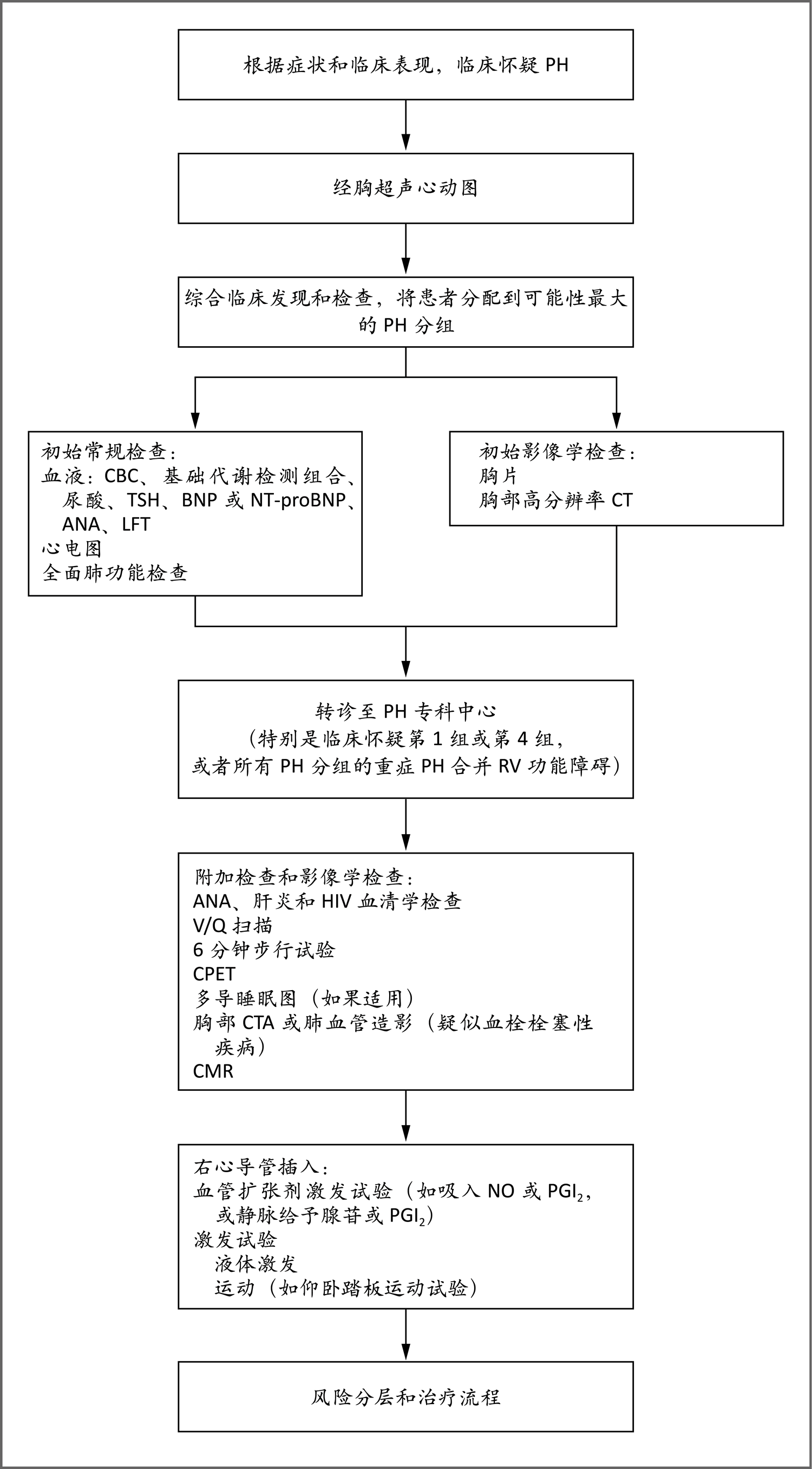
当根据症状和体征怀疑PH时,经胸超声心动图是全科医师开出的最重要筛查项目。临床评估中可综合其他血液和影像学检查,以确定诊断PH的最佳方法,以及可能的亚组或者相关状况(第1组)或其他疾病(如第2~5组)。怀疑患第1组(PAH),第4组(慢性血栓栓塞性PH)或其他形式重度PH合并右心室(RV)功能障碍(超声心动图)的患者通常应转诊至专科PH中心,他们在专科中心接受附加检查、影像学检查和右心导管检查,以进一步确定PH的性质和严重程度。多导睡眠图有助于排除可能导致或加重PH的相关睡眠障碍。吸入一氧化氮(NO)或吸入前列环素(PGI2)类似物或者静脉注射腺苷或PGI2类似物的血管扩张剂激发试验通常仅用于特发性PAH、遗传性PAH或者与药物或毒素相关的PAH患者60。液体激发试验或运动激发试验分别用于检测左心疾病或运动诱发的PH。心肺运动试验(CPET)有助于识别呼吸困难的心源和肺源原因,并测定指标(如最大耗氧量或峰值),这些指标可被纳入欧洲心脏病学会-欧洲呼吸学会(European Society of Cardiology-European Respiratory Society,ESC-ERS)风险分层(有关ESC-ERS风险评分和REVEAL[评估早期和长期PAH疾病治疗的登记系统,Registry to Evaluate Early and Long-Term PAH Disease Management]评分的详细信息,见补充附录图S4)。此流程改编自Galiè等60的论文。ANA表示抗核抗体,BNP表示B型利钠肽,CBC表示全血细胞计数,CMR表示心脏磁共振,LFT表示肝功能检查,NT-proBNP表示氨基末端proBNP,TSH表示促甲状腺素。
胸片可提示心脏扩大和肺动脉扩张,以及肺实质或胸壁异常。应常规进行胸部计算机断层扫描(CT),以排除肺实质疾病。通气-灌注扫描在临床流程中仍然必不可少,因为如果灌注正常,则不太可能是慢性血栓栓塞性肺高压(CTEPH)。胸部CT血管造影虽然被认为不如通气-灌注扫描灵敏,但前者可能揭示慢性血栓栓塞性疾病征象,如既往血栓造成的充盈缺损或者楔形或不规则线状阴影。它还有助于确定病变的手术可及性,并且可排除其他诊断(如肺动脉狭窄或肿瘤及纤维性纵隔炎)。
肺功能检查可提示阻塞性或限制性肺疾病。肺部对一氧化碳的单次呼吸弥散量(肺动脉高压患者通常降低)可以与其他临床表现和TTE表现(右心房扩大和三尖瓣反流速度)一起被纳入循证流程,用于检测无症状硬皮病谱系疾病患者的肺动脉高压62。心电图对于寻找心房或心室肥厚、缺血性心脏病或心律失常证据很重要。
心脏MRI(图S3)是评估右心室的标准方法,因为它可准确测量心腔解剖结构、容积、质量、功能和流量,以及心肌灌注38。虽然尚未普及,但在专科中心,心脏MRI已越来越多地用于诊断和治疗肺高压。仍然仅用于科研的其他先进影像学技术包括三维超声心动图、四维血流MRI和正电子发射断层扫描,它们可帮助我们了解右心室代谢活动63。
血流动力学
诊断肺动脉高压需要插入右心导管,以便直接评估肺血流动力学和心输出量,以及计算肺血管阻力。这是采取治疗前,诊断流程中的必要步骤(图3)。它对于确认肺高压(毛细血管前、毛细血管后或联合)及其类型至关重要,并为风险分层提供必要指标。结构化的临床评估(图3)有助于将患者分配到具体的肺高压分组(图1),这对于确定适当的治疗方案至关重要,不过我们日益认识到,任何患者都可能属于不止一个分组。
风险分层
风险评估的重要性在特发性肺动脉高压研究(当时的重点主要是基线血流动力学)的早期已经认识到64。2015年欧洲心脏病学会-欧洲呼吸学会(ESC-ERS)肺动脉高压诊疗指南65强调,在基线和随访时,结合临床、功能和血流动力学指标,将患者分层到低危、中危和高危人群是一个越来越重要的优先事项,而且也是选择治疗方案的工具。在对肺动脉高压登记系统67-69数据进行的回顾性分析和对一项大规模前瞻性临床试验70数据进行的事后分析中,ESC-ERS“风险表”和其他风险评分(如REVEAL风险评分计算器66](图S4)已成功地用于评估生存率。机器学习算法提高了分层方法的预测能力,揭示了多个危险因素间相互依赖的动态影响,从而避免了有限临床指标与特定结局具有独立关系这一假设71。
治疗
在靶向治疗出现之前的治疗方案中,基本支持措施是固定组成部分,其中包括利尿剂(以达到正常血容量),以及在休息、睡眠或运动时根据需要吸氧(以维持足够的血红蛋白氧饱和度)(图4)。睡眠障碍性呼吸(可并发任何类型的心肺疾病)在毛细血管前肺高压72患者中很普遍,应做出诊断并在适合的情况下采取治疗。抗凝治疗(根据回顾性分析显示的生存获益,之前曾推荐抗凝治疗73,74)现在仅推荐用于特发性肺动脉高压(根据欧洲登记系统的数据,不推荐用于其他类型的肺动脉高压)75,以及根据每个病例的具体情况和风险-获益分析结果,用于第4组肺高压(CTEPH)(凝血增加是此类患者的主要问题)。根据对照试验的荟萃分析结果,建议患者进行可耐受的心肺运动76(图4)。患者应及时接种疫苗。虽然临床上第一次描述肺高压与肺动脉高压的第一种有效治疗药物获批(根据在“原发性肺高压”患者中对前列环素进行的随机、非安慰剂对照试验77)相隔了40年,但过去20年间有一系列临床试验靶向肺动脉高压的三个信号通路78(图5)。这些试验确立了肺动脉高压(第1组)和CTEPH(第4组)的现行靶向疗法。试验结果不适用于WSPH分类中的其他分组,只有曲前列尼尔吸入剂是例外,根据近期一项随机、3期临床试验的结果,美国食品药品管理局(FDA)已批准曲前列尼尔用于治疗间质性肺病相关肺高压(第3组)79。CTEPH评估最好在专科中心进行,并且这些中心应首先考虑通过肺动脉内膜切除术实施确定性治疗。对于CTEPH不能手术或动脉内膜切除术后残留肺动脉高压的患者,可考虑药物治疗、肺球囊血管成形术或两者兼用。
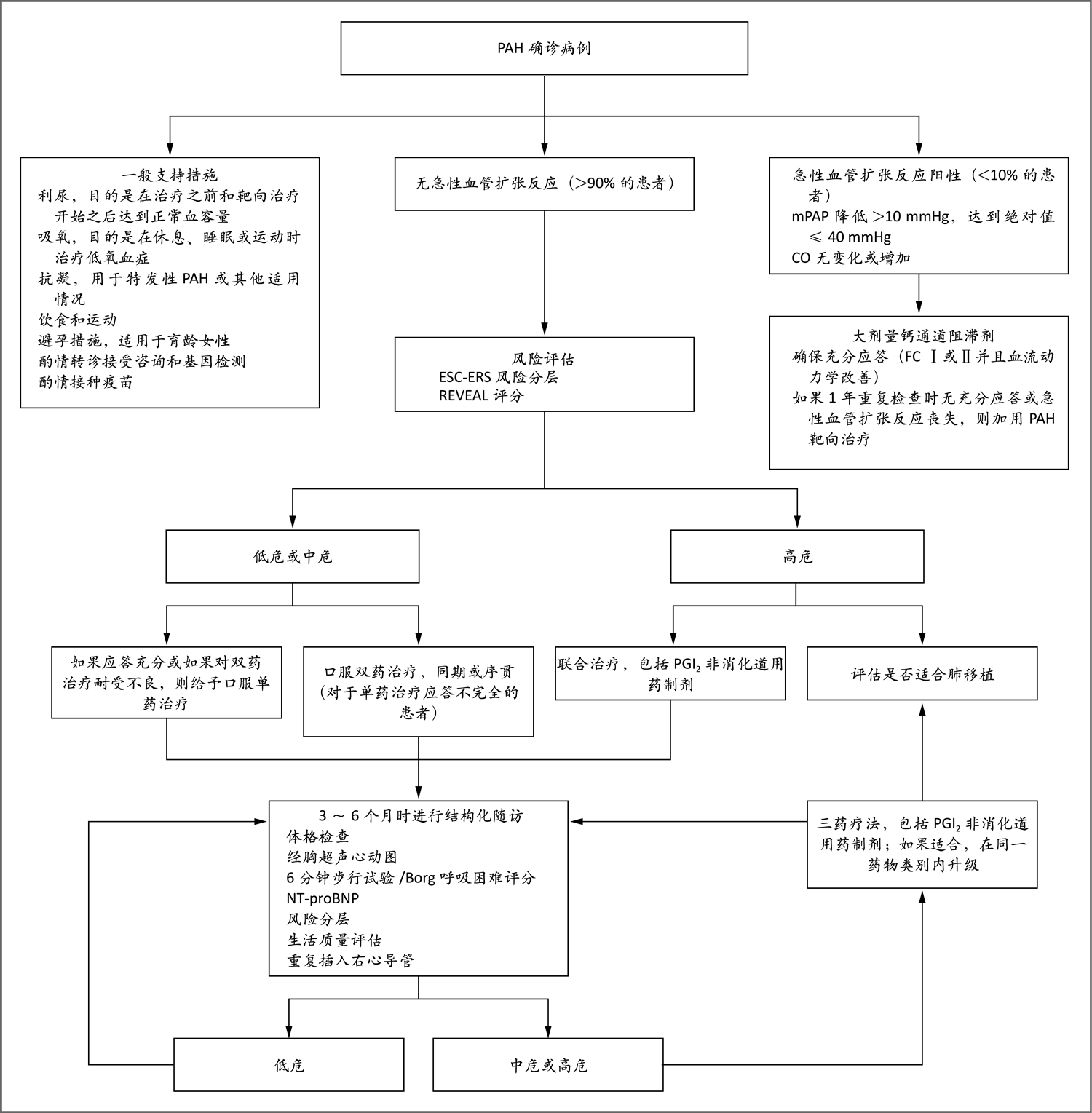
此流程改编自Galiè等60的论文。有关ESC-ERS风险评分和REVEAL评分的详细信息见图S4。Borg呼吸困难量表用于评估患者在6分钟步行试验期间报告的呼吸困难;量表评分范围为0~10分,评分较高表示呼吸困难较严重。对于推荐三药治疗的患者,一般应在结构化随访时评估其是否适合肺移植。FC表示功能分类。
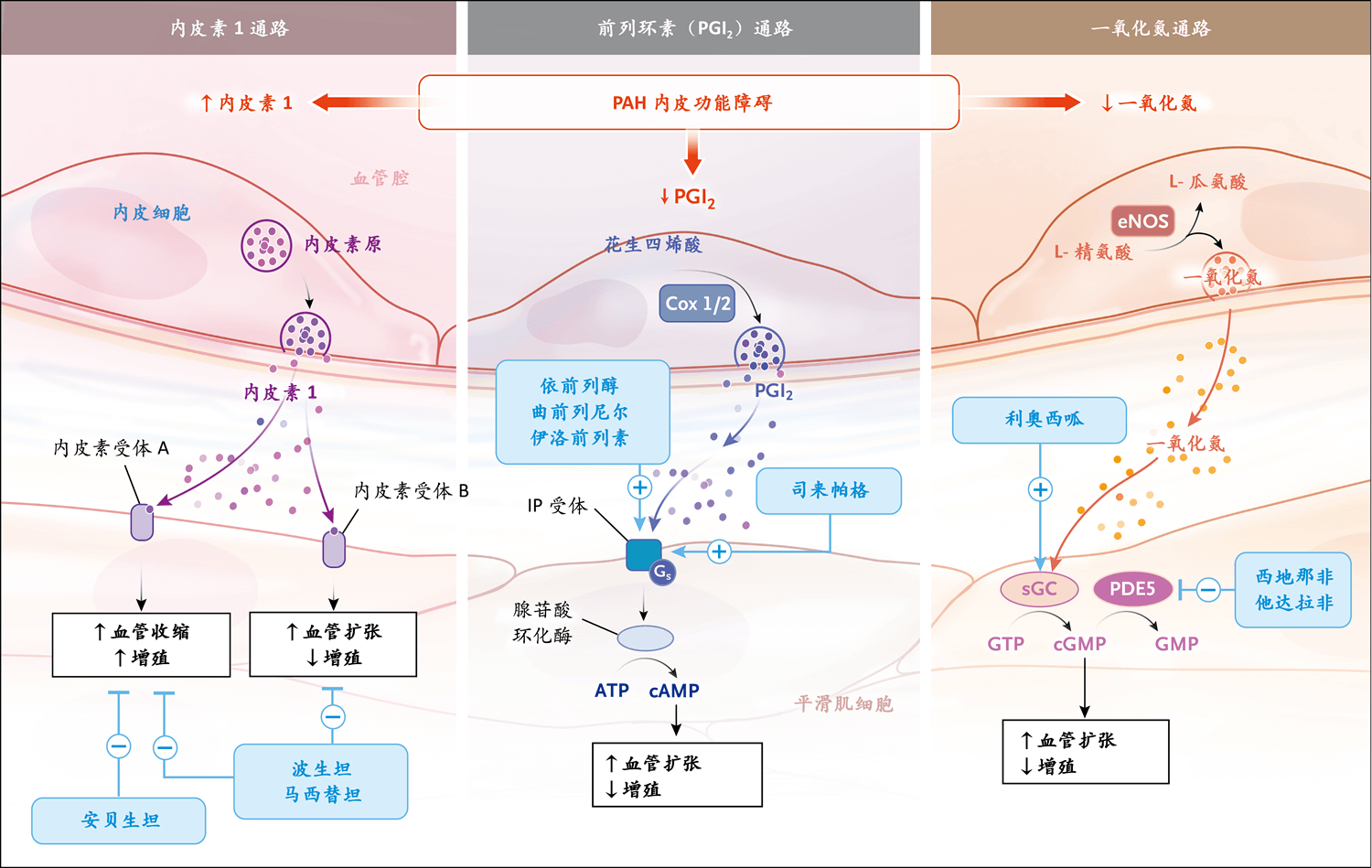
目前的靶向疗法旨在通过抑制内皮素通路和增强前列环素(PGI2)和NO通路来纠正内皮功能障碍27。PAH患者增加的内皮素1(ET1)可与内皮素A(ETA)受体结合,引起血管收缩(平滑肌细胞)和细胞增殖,也可与内皮素B(ETB)受体结合,引起血管扩张和抗增殖。因此,目前有双重ETA-ETB受体拮抗剂(如波生坦和马西替坦)或选择性ETA受体拮抗剂(如安贝生坦),这样ETB受体可发挥功能。在PAH中,PGI2和NO通路的表达和功能降低,分别导致环磷酸腺苷(cAMP)和环磷酸鸟苷(cGMP)减少,而cAMP和cGMP是负责血管扩张和抗增殖的第二信使。可增加cAMP的药物包括静脉给药(如依前列醇和曲前列尼尔)、皮下给药(曲前列尼尔)、吸入给药(如伊洛前列素和曲前列尼尔)、口服给药(曲前列尼尔)的PGI2类似物,或者使用口服给药的PGI2受体(IP)激动剂(如司来帕格)。可增加cGMP释放的方式包括吸入NO(基本上用于心导管室或重症监护病房,NO可刺激可溶性鸟苷酸环化酶[sGC]),或者使用口服PDE5抑制剂(西地那非或他达拉非)抑制5型磷酸二酯酶(PDE5,PDE5可将cGMP降解为GMP)。直接sGC刺激剂(如口服利奥西呱)可增加cGMP释放(与NO释放无关)。这些药物已被美国食品药品管理局批准用于mPAP≥25 mmHg的PAH患者。这些药物的不良事件通常可接受,但有由于血管扩张作用引起的相同副作用,包括头痛和头晕(尤其是在治疗开始时)、面部潮红和上呼吸道充血,体循环低血压(全身给药时更常见,使用某些需要缓慢增加剂量的药物时也常见,如利奥西呱和司来帕格)、消化系统症状(如腹胀、恶心或呕吐和腹泻)和皮疹(PGI2类似物)。液体潴留(如足部水肿)是治疗开始时的常见问题,需要调整利尿剂的剂量。其他特异性副作用包括咳嗽(如吸入曲前列尼尔)、转氨酶水平升高(波生坦)、贫血(马西替坦)、视力改变(PDE5抑制剂,但非常罕见)、皮肤刺激或蜂窝织炎(皮下给药的曲前列尼尔)、血小板减少或骨髓抑制(全身给药的PGI2类似物)。AA表示花生四烯酸,AC表示腺苷酸环化酶,Cox 1/2表示环加氧酶1/2,eNOS表示内皮型一氧化氮合酶,GS表示G蛋白偶联受体,GTP表示鸟苷三磷酸。
随机对照试验(RCT)的设计在近10年间发生了巨大变化。6分钟步行距离是大多数早期RCT(研究期通常为12周)的主要终点。然而,有多方呼吁设立相关性更高的终点 80,因此RCT转向了复合终点,包括住院、肺动脉高压恶化、死亡率和治疗方案升级。RCT设计发生的另外一个重大变化是评估新疗法时,研究者在肺动脉高压基础疗法之上加用新疗法,或者采用同期联合治疗,而不采用单药治疗。上述变化需要纳入更多患者,开展更长时间才能达到主要结局。例如,SERAPHIN(应用内皮素受体拮抗剂改善肺动脉高压患者临床结局的研究,Study with an Endothelin Receptor Antagonist in Pulmonary Arterial Hypertension to Improve Clinical Outcome)。SERAPHIN的研究人群是接受安慰剂或基础治疗(吸入或口服药物,不包括其他内皮素受体拮抗剂)的700多例有症状肺动脉高压患者,该研究显示了马西替坦(双重内皮素受体拮抗剂)在减少复合主要终点首次发生方面的疗效81。之后的一项RCT AMBITION(安贝生坦和他达拉非治疗肺动脉高压患者,Ambrisentan and Tadalafil in Patients with Pulmonary Arterial Hypertension)试验在既往未接受过肺动脉高压治疗的患者中比较了FDA批准的两种药物(安贝生坦和他达拉非)同期联合治疗和各药单药治疗82。与任一药物单药治疗相比,联合治疗的主要终点(至事件发生时间分析中的首起临床失败事件)风险降低。然而,以上两项大规模试验中的主要终点都是主要来自住院率降低(住院的主要原因是肺动脉高压恶化)(这是一项有临床意义的结局),因为因右心室衰竭(肺动脉高压患者住院的主要原因)住院预示着预后很差83。
虽然近期大多数大型RCT均显示联合口服治疗对生存期无影响81,82,84,但对评估肺动脉高压疗法的早期RCT(平均持续时间12~16周)进行的大型荟萃分析显示,与安慰剂相比,治疗显著降低了死亡率85,这与从大型登记系统获得的数据一致86,87。
用于指导治疗的治疗流程(包括肺动脉高压各种已获批疗法的推荐级别和证据水平)可参考综合指南65。根据严格标准(平均肺动脉压降低≥10 mmHg,达到绝对值≤40 mmHg,伴有心输出量增加或无变化),有肺血管扩张反应(一般在最初插入右心导管期间对吸入一氧化氮有反应)的患者可接受大剂量钙通道阻滞剂单药治疗,前提是单药治疗使患者在治疗至少1年后的重复检查时达到纽约心脏学会(NYHA)心功能分级Ⅰ级或Ⅱ级,并且维持血流动力学改善1(不到10%的特发性肺动脉高压患者可达到这一点88)。如果出现临床病情恶化或血管扩张反应丧失,则应根据公认的治疗流程加用肺动脉高压特异性治疗。
单药治疗可用于急性血管扩张反应阳性的患者以及以下患者:既往应答良好的患者(NYHA心功能分级为Ⅰ级或Ⅱ级,并且维持血流动力学改善),有左心疾病重要危险因素(如体循环高血压、冠心病或心房颤动)的老年患者(>75岁),疑似患肺静脉闭塞性疾病或肺毛细血管瘤病的患者,病情很轻的患者(NYHA心功能分级为Ⅰ级,肺血管阻力为3~4 WU,超声心动图显示右心室功能正常),以及联合治疗有不可接受的副作用的患者60。除此之外,大多数肺动脉高压患者目前接受两种口服药物同期联合治疗(适合情况下,在同一药物类别内提高剂量)或者接受序贯联合治疗。如果药物治疗不能将风险降低至低水平或中等水平,则建议将患者转诊,评估是否适合接受肺移植。同期三药联合治疗对肺动脉高压患者的作用尚不清楚。
对于终末期肺动脉高压或等待肺移植的患者,偶尔考虑心房间隔造口术。心房间隔造口术的优点是可降低右心房和右心室负荷,延缓右心室衰竭,同时改善左心室前负荷和心输出量,但代价是右向左分流导致氧合减少。
未来方向
日益高精的计算能力,结合先进的蛋白质组学平台89或影像学检查90,产生了机器学习技术。我们可应用这些技术为肺动脉高压开发出有前景且强大的诊断工具。目前的趋势是对生物标志物和各种其他“组学”(蛋白质组学和基因组学)开展大规模研究,这应该有助于确定适用于肺高压各分组的特定机制通路(肺高压各分组的共同和独特通路);这一趋势还将产生精准医学(其中考虑基因、环境和生活方式等因素),这一过程类似于当前癌症疗法的产生过程。然而,要取得成功,我们需要在国家和国际水平进行各中心间密切合作,从而创建大型登记系统(对于临床和影像学表型),以及组织、生物标志物、遗传学和蛋白质组学的生物样本库。这一点对于肺动脉高压这样的罕见综合征尤其重要。如蛋白质组学研究91和基因组学研究59所示,将分子分类纳入目前的分类已经是一个现实的目标。
根据免疫失调可能引发或促进肺动脉高压发病的观点,针对特定免疫通路的临床试验最近已经启动。靶向硬皮病相关肺动脉高压患者的B细胞似乎对生物标志物机器学习方法分析确定的一个患者亚组有效,这提示该疗法可能可以用于这一疾病的辅助免疫治疗92。同样,人们仍然十分关注靶向发生改变的生长因子信号传导,尽管酪氨酸激酶抑制剂伊马替尼的情况令人持谨慎态度,该药物在2期试验中取得了令人鼓舞的结果,但3期试验中出现了严重副作用(如硬膜下出血),因而FDA未批准伊马替尼用于治疗肺动脉高压93。
最后,关于钙调磷酸酶抑制剂FK506(FK506可上调BMPR-Ⅱ表达水平55)的一项试验94引人关注,因为挽救BMPR-Ⅱ信号通路在对抗促增殖和促炎症TGF-β通路方面具有重要意义。同样,近期一项2期临床试验表明,在接受基础治疗的肺动脉高压患者中,sotatercept(设计用于结合TGF-β配体激活蛋白的首创融合蛋白)降低了肺血管阻力和血清氨基末端前B型利钠肽水平,并改善了功能95。这一非常有前景的新疗法目前正在开展3期临床试验。
作者信息
Paul M. Hassoun, M.D.
From the Division of Pulmonary and Critical Care Medicine, Department of Medicine, Johns Hopkins University School of Medicine, Baltimore. Dr. Hassoun can be contacted at phassoun@jhmi.edu or at the Division of Pulmonary and Critical Care Medicine, Johns Hopkins University School of Medicine, 1830 E. Monument St., Baltimore, MD 21287.
参考文献
1. Simonneau G, Montani D, Celermajer DS, et al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J 2019;53:1801913-1801913.
2. von Romberg E. Über sklerose der lungenarterie. Dtsch Arch Klin Med 1891;48:197-206.
3. Forssmann W. Die sondierung des rechten herzens. Klin Wochenschr 1929;8:2085-2087.
4. Richards DW. The contributions of right heart catheterization to physiology and medicine, with some observations on the physiopathology of pulmonary heart disease. Am Heart J 1957;54:161-171.
5. Dresdale DT, Schultz M, Michtom RJ. Primary pulmonary hypertension. I. Clinical and hemodynamic study. Am J Med 1951;11:686-705.
6. Kay JM, Smith P, Heath D. Aminorex and the pulmonary circulation. Thorax 1971;26:262-270.
7. Hatano S, Strasser T, eds. Primary pulmonary hypertension: report on a WHO meeting, Geneva, 15–17 October, 1973. Geneva: World Health Organization, 1975.
8. Lane KB, Machado RD, Pauciulo MW, et al. Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension. Nat Genet 2000;26:81-84.
9. Deng Z, Morse JH, Slager SL, et al. Familial primary pulmonary hypertension (gene PPH1) is caused by mutations in the bone morphogenetic protein receptor-II gene. Am J Hum Genet 2000;67:737-744.
10. Humbert M, Sitbon O, Chaouat A, et al. Pulmonary arterial hypertension in France: results from a national registry. Am J Respir Crit Care Med 2006;173:1023-1030.
11. Hoeper MM, Huscher D, Ghofrani HA, et al. Elderly patients diagnosed with idiopathic pulmonary arterial hypertension: results from the COMPERA registry. Int J Cardiol 2013;168:871-880.
12. Rich S, Haworth SG, Hassoun PM, Yacoub MH. Pulmonary hypertension: the unaddressed global health burden. Lancet Respir Med 2018;6:577-579.
13. Wu W, He J, Shao X. Incidence and mortality trend of congenital heart disease at the global, regional, and national level, 1990–2017. Medicine (Baltimore) 2020;99(23):e20593-e20593.
14. Hoeper MM, Humbert M, Souza R, et al. A global view of pulmonary hypertension. Lancet Respir Med 2016;4:306-322.
15. Gorter TM, Hoendermis ES, van Veldhuisen DJ, et al. Right ventricular dysfunction in heart failure with preserved ejection fraction: a systematic review and meta-analysis. Eur J Heart Fail 2016;18:1472-1487.
16. Miller WL, Grill DE, Borlaug BA. Clinical features, hemodynamics, and outcomes of pulmonary hypertension due to chronic heart failure with reduced ejection fraction: pulmonary hypertension and heart failure. JACC Heart Fail 2013;1:290-299.
17. Moore LG, Niermeyer S, Zamudio S. Human adaptation to high altitude: regional and life-cycle perspectives. Am J Phys Anthropol 1998;107:Suppl 27:25-64.
18. Deol AK, Fleming FM, Calvo-Urbano B, et al. Schistosomiasis — assessing progress toward the 2020 and 2025 global goals. N Engl J Med 2019;381:2519-2528.
19. GBD 2017 HIV collaborators. Global, regional, and national incidence, prevalence, and mortality of HIV, 1980–2017, and forecasts to 2030, for 195 countries and territories: a systematic analysis for the Global Burden of Diseases, Injuries, and Risk Factors Study 2017. Lancet HIV 2019;6(12):e831-e859.
20. Sikirica M, Iorga SR, Bancroft T, Potash J. The economic burden of pulmonary arterial hypertension (PAH) in the US on payers and patients. BMC Health Serv Res 2014;14:676-676.
21. Southgate L, Machado RD, Gräf S, Morrell NW. Molecular genetic framework underlying pulmonary arterial hypertension. Nat Rev Cardiol 2020;17:85-95.
22. Wagenvoort CA. The pathology of primary pulmonary hypertension. J Pathol 1970;101:Pi-Pi.
23. Price LC, Wort SJ, Perros F, et al. Inflammation in pulmonary arterial hypertension. Chest 2012;141:210-221.
24. Launay D, Sobanski V, Hachulla E, Humbert M. Pulmonary hypertension in systemic sclerosis: different phenotypes. Eur Respir Rev 2017;26:170056-170056.
25. Gerges C, Gerges M, Friewald R, et al. Microvascular disease in chronic thromboembolic pulmonary hypertension: hemodynamic phenotyping and histomorphometric assessment. Circulation 2020;141:376-386.
26. Fayyaz AU, Edwards WD, Maleszewski JJ, et al. Global pulmonary vascular remodeling in pulmonary hypertension associated with heart failure and preserved or reduced ejection fraction. Circulation 2018;137:1796-1810.
27. Budhiraja R, Tuder RM, Hassoun PM. Endothelial dysfunction in pulmonary hypertension. Circulation 2004;109:159-165.
28. Galambos C, Sims-Lucas S, Abman SH, Cool CD. Intrapulmonary bronchopulmonary anastomoses and plexiform lesions in idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med 2016;193:574-576.
29. Perros F, Dorfmüller P, Montani D, et al. Pulmonary lymphoid neogenesis in idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med 2012;185:311-321.
30. Soon E, Holmes AM, Treacy CM, et al. Elevated levels of inflammatory cytokines predict survival in idiopathic and familial pulmonary arterial hypertension. Circulation 2010;122:920-927.
31. Hassoun PM, Mouthon L, Barberà JA, et al. Inflammation, growth factors, and pulmonary vascular remodeling. J Am Coll Cardiol 2009;54:Suppl 1:S10-S19.
32. Vonk Noordegraaf A, Chin KM, Haddad F, et al. Pathophysiology of the right ventricle and of the pulmonary circulation in pulmonary hypertension: an update. Eur Respir J 2019;53:1801900-1801900.
33. Ryan JJ, Archer SL. The right ventricle in pulmonary arterial hypertension: disorders of metabolism, angiogenesis and adrenergic signaling in right ventricular failure. Circ Res 2014;115:176-188.
34. Vanderpool RR, Pinsky MR, Naeije R, et al. RV-pulmonary arterial coupling predicts outcome in patients referred for pulmonary hypertension. Heart 2015;101:37-43.
35. Trip P, Kind T, van de Veerdonk MC, et al. Accurate assessment of load-independent right ventricular systolic function in patients with pulmonary hypertension. J Heart Lung Transplant 2013;32:50-55.
36. Sanz J, Kariisa M, Dellegrottaglie S, et al. Evaluation of pulmonary artery stiffness in pulmonary hypertension with cardiac magnetic resonance. JACC Cardiovasc Imaging 2009;2:286-295.
37. Nie L, Li J, Zhang S, et al. Correlation between right ventricular-pulmonary artery coupling and the prognosis of patients with pulmonary arterial hypertension. Medicine (Baltimore) 2019;98(40):e17369-e17369.
38. Lahm T, Douglas IS, Archer SL, et al. Assessment of right ventricular function in the research setting: knowledge gaps and pathways forward. An official American Thoracic Society research statement. Am J Respir Crit Care Med 2018;198(4):e15-e43.
39. Kawut SM, Lima JAC, Barr RG, et al. Sex and race differences in right ventricular structure and function: the multi-ethnic study of atherosclerosis-right ventricle study. Circulation 2011;123:2542-2551.
40. Ventetuolo CE, Ouyang P, Bluemke DA, et al. Sex hormones are associated with right ventricular structure and function: the MESA-right ventricle study. Am J Respir Crit Care Med 2011;183:659-667.
41. Jacobs W, van de Veerdonk MC, Trip P, et al. The right ventricle explains sex differences in survival in idiopathic pulmonary arterial hypertension. Chest 2014;145:1230-1236.
42. Rain S, da Silva Goncalves Bos D, Handoko ML, et al. Protein changes contributing to right ventricular cardiomyocyte diastolic dysfunction in pulmonary arterial hypertension. J Am Heart Assoc 2014;3(3):e000716-e000716.
43. Hsu S, Kokkonen-Simon KM, Kirk JA, et al. Right ventricular myofilament functional differences in humans with systemic sclerosis-associated versus idiopathic pulmonary arterial hypertension. Circulation 2018;137:2360-2370.
44. Trembath RC, Thomson JR, Machado RD, et al. Clinical and molecular genetic features of pulmonary hypertension in patients with hereditary hemorrhagic telangiectasia. N Engl J Med 2001;345:325-334.
45. Harrison RE, Berger R, Haworth SG, et al. Transforming growth factor-beta receptor mutations and pulmonary arterial hypertension in childhood. Circulation 2005;111:435-441.
46. Nasim MT, Ogo T, Ahmed M, et al. Molecular genetic characterization of SMAD signaling molecules in pulmonary arterial hypertension. Hum Mutat 2011;32:1385-1389.
47. Gräf S, Haimel M, Bleda M, et al. Identification of rare sequence variation underlying heritable pulmonary arterial hypertension. Nat Commun 2018;9:1416-1416.
48. Austin ED, Ma L, LeDuc C, et al. Whole exome sequencing to identify a novel gene (caveolin-1) associated with human pulmonary arterial hypertension. Circ Cardiovasc Genet 2012;5:336-343.
49. Ma L, Roman-Campos D, Austin ED, et al. A novel channelopathy in pulmonary arterial hypertension. N Engl J Med 2013;369:351-361.
50. Kerstjens-Frederikse WS, Bongers EMHF, Roofthooft MTR, et al. TBX4 mutations (small patella syndrome) are associated with childhood-onset pulmonary arterial hypertension. J Med Genet 2013;50:500-506.
51. Zhu N, Welch CL, Wang J, et al. Rare variants in SOX17 are associated with pulmonary arterial hypertension with congenital heart disease. Genome Med 2018;10:56-56.
52. Best DH, Sumner KL, Austin ED, et al. EIF2AK4 mutations in pulmonary capillary hemangiomatosis. Chest 2014;145:231-236.
53. Eyries M, Montani D, Girerd B, et al. EIF2AK4 mutations cause pulmonary veno-occlusive disease, a recessive form of pulmonary hypertension. Nat Genet 2014;46:65-69.
54. Potus F, Pauciulo MW, Cook EK, et al. Novel mutations and decreased expression of the epigenetic regulator TET2 in pulmonary arterial hypertension. Circulation 2020;141:1986-2000.
55. Spiekerkoetter E, Tian X, Cai J, et al. FK506 activates BMPR2, rescues endothelial dysfunction, and reverses pulmonary hypertension. J Clin Invest 2013;123:3600-3613.
56. Long L, Ormiston ML, Yang X, et al. Selective enhancement of endothelial BMPR-II with BMP9 reverses pulmonary arterial hypertension. Nat Med 2015;21:777-785.
57. Morrell NW, Aldred MA, Chung WK, et al. Genetics and genomics of pulmonary arterial hypertension. Eur Respir J 2019;53:1801899-1801899.
58. Zhu N, Swietlik EM, Welch CL, et al. Rare variant analysis of 4241 pulmonary arterial hypertension cases from an international consortium implicates FBLN2, PDGFD, and rare de novo variants in PAH. Genome Med 2021;13:80-80.
59. Hemnes AR, Beck GJ, Newman JH, et al. PVDOMICS: a multi-center study to improve understanding of pulmonary vascular disease through phenomics. Circ Res 2017;121:1136-1139.
60. Galiè N, Channick RN, Frantz RP, et al. Risk stratification and medical therapy of pulmonary arterial hypertension. Eur Respir J 2019;53:1801889-1801889.
61. Fijalkowska A, Kurzyna M, Torbicki A, et al. Serum N-terminal brain natriuretic peptide as a prognostic parameter in patients with pulmonary hypertension. Chest 2006;129:1313-1321.
62. Coghlan JG, Denton CP, Grünig E, et al. Evidence-based detection of pulmonary arterial hypertension in systemic sclerosis: the DETECT study. Ann Rheum Dis 2014;73:1340-1349.
63. Rengier F, Melzig C, Derlin T, Marra AM, Vogel-Claussen J. Advanced imaging in pulmonary hypertension: emerging techniques and applications. Int J Cardiovasc Imaging 2019;35:1407-1420.
64. D’Alonzo GE, Barst RJ, Ayres SM, et al. Survival in patients with primary pulmonary hypertension. Results from a national prospective registry. Ann Intern Med 1991;115:343-349.
65. Galiè N, Humbert M, Vachiery JL, et al. 2015 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension: the Joint Task Force for the diagnosis and treatment of pulmonary hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS). Eur Respir J 2015;46:903-975.
66. Benza RL, Farber HW, Frost A, et al. REVEAL risk scores applied to riociguat-treated patients in PATENT-2: impact of changes in risk score on survival. J Heart Lung Transplant 2018;37:513-519.
67. Hoeper MM, Kramer T, Pan Z, et al. Mortality in pulmonary arterial hypertension: prediction by the 2015 European pulmonary hypertension guidelines risk stratification model. Eur Respir J 2017;50:1700740-1700740.
68. Kylhammar D, Kjellström B, Hjalmarsson C, et al. A comprehensive risk stratification at early follow-up determines prognosis in pulmonary arterial hypertension. Eur Heart J 2018;39:4175-4181.
69. Boucly A, Weatherald J, Savale L, et al. Risk assessment, prognosis and guideline implementation in pulmonary arterial hypertension. Eur Respir J 2017;50:1700889-1700889.
70. Galiè N, Jansa P, Pulido T, et al. SERAPHIN haemodynamic substudy: the effect of the dual endothelin receptor antagonist macitentan on haemodynamic parameters and NT-proBNP levels and their association with disease progression in patients with pulmonary arterial hypertension. Eur Heart J 2017;38:1147-1155.
71. Kanwar MK, Gomberg-Maitland M, Hoeper M, et al. Risk stratification in pulmonary arterial hypertension using Bayesian analysis. Eur Respir J 2020;56:2000008-2000008.
72. Thurnheer R, Ulrich S, Bloch KE. Precapillary pulmonary hypertension and sleep-disordered breathing: is there a link? Respiration 2017;93:65-77.
73. Rich S, Kaufmann E, Levy PS. The effect of high doses of calcium-channel blockers on survival in primary pulmonary hypertension. N Engl J Med 1992;327:76-81.
74. Fuster V, Steele PM, Edwards WD, Gersh BJ, McGoon MD, Frye RL. Primary pulmonary hypertension: natural history and the importance of thrombosis. Circulation 1984;70:580-587.
75. Olsson KM, Delcroix M, Ghofrani HA, et al. Anticoagulation and survival in pulmonary arterial hypertension: results from the Comparative, Prospective Registry of Newly Initiated Therapies for Pulmonary Hypertension (COMPERA). Circulation 2014;129:57-65.
76. Buys R, Avila A, Cornelissen VA. Exercise training improves physical fitness in patients with pulmonary arterial hypertension: a systematic review and meta-analysis of controlled trials. BMC Pulm Med 2015;15:40-40.
77. Barst RJ, Rubin LJ, Long WA, et al. A comparison of continuous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension. N Engl J Med 1996;334:296-301.
78. Humbert M, Lau EM, Montani D, Jaïs X, Sitbon O, Simonneau G. Advances in therapeutic interventions for patients with pulmonary arterial hypertension. Circulation 2014;130:2189-2208.
79. Waxman A, Restrepo-Jaramillo R, Thenappan T, et al. Inhaled treprostinil in pulmonary hypertension due to interstitial lung disease. N Engl J Med 2021;384:325-334.
80. Gaine S, Simonneau G. The need to move from 6-minute walk distance to outcome trials in pulmonary arterial hypertension. Eur Respir Rev 2013;22:487-494.
81. Pulido T, Adzerikho I, Channick RN, et al. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N Engl J Med 2013;369:809-818.
82. Galiè N, Barberà JA, Frost AE, et al. Initial use of ambrisentan plus tadalafil in pulmonary arterial hypertension. N Engl J Med 2015;373:834-844.
83. Campo A, Mathai SC, Le Pavec J, et al. Outcomes of hospitalisation for right heart failure in pulmonary arterial hypertension. Eur Respir J 2011;38:359-367.
84. Sitbon O, Channick R, Chin KM, et al. Selexipag for the treatment of pulmonary arterial hypertension. N Engl J Med 2015;373:2522-2533.
85. Galiè N, Manes A, Negro L, Palazzini M, Bacchi-Reggiani ML, Branzi A. A meta-analysis of randomized controlled trials in pulmonary arterial hypertension. Eur Heart J 2009;30:394-403.
86. Humbert M, Sitbon O, Chaouat A, et al. Survival in patients with idiopathic, familial, and anorexigen-associated pulmonary arterial hypertension in the modern management era. Circulation 2010;122:156-163.
87. Benza RL, Miller DP, Barst RJ, Badesch DB, Frost AE, McGoon MD. An evaluation of long-term survival from time of diagnosis in pulmonary arterial hypertension from the REVEAL Registry. Chest 2012;142:448-456.
88. Sitbon O, Humbert M, Jaïs X, et al. Long-term response to calcium channel blockers in idiopathic pulmonary arterial hypertension. Circulation 2005;111:3105-3111.
89. Sweatt AJ, Hedlin HK, Balasubramanian V, et al. Discovery of distinct immune phenotypes using machine learning in pulmonary arterial hypertension. Circ Res 2019;124:904-919.
90. Swift AJ, Lu H, Uthoff J, et al. A machine learning cardiac magnetic resonance approach to extract disease features and automate pulmonary arterial hypertension diagnosis. Eur Heart J Cardiovasc Imaging 2021;22:236-245.
91. Rhodes CJ, Wharton J, Ghataorhe P, et al. Plasma proteome analysis in patients with pulmonary arterial hypertension: an observational cohort study. Lancet Respir Med 2017;5:717-726.
92. Zamanian RT, Badesch D, Chung L, et al. Safety and efficacy of B-cell depletion with rituximab for the treatment of systemic sclerosis-associated pulmonary arterial hypertension: a multicenter, double-blind, randomized, placebo-controlled trial. Am J Respir Crit Care Med 2021;204:209-221.
93. Hoeper MM, Barst RJ, Bourge RC, et al. Imatinib mesylate as add-on therapy for pulmonary arterial hypertension: results of the randomized IMPRES study. Circulation 2013;127:1128-1138.
94. Spiekerkoetter E, Sung YK, Sudheendra D, et al. Randomised placebo-controlled safety and tolerability trial of FK506 (tacrolimus) for pulmonary arterial hypertension. Eur Respir J 2017;50:1602449-1602449.
95. Humbert M, McLaughlin V, Gibbs JSR, et al. Sotatercept for the treatment of pulmonary arterial hypertension. N Engl J Med 2021;384:1204-1215.
特别申明:本文为转载文章,转载自nejm医学前沿,不代表贪吃的夜猫子立场,如若转载,请注明出处:http://www.nejmqianyan.cn/article-info?permalinks=YXQYra2000348
 微信扫一扫
微信扫一扫  支付宝扫一扫
支付宝扫一扫 




