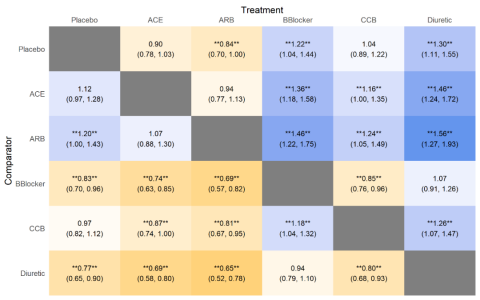
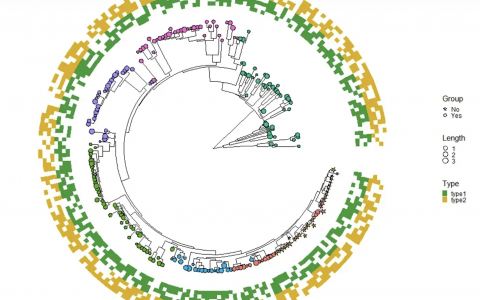
本期分享的期刊是:Nature Microbiology上面的Figure 1,具体文章信息如下图:

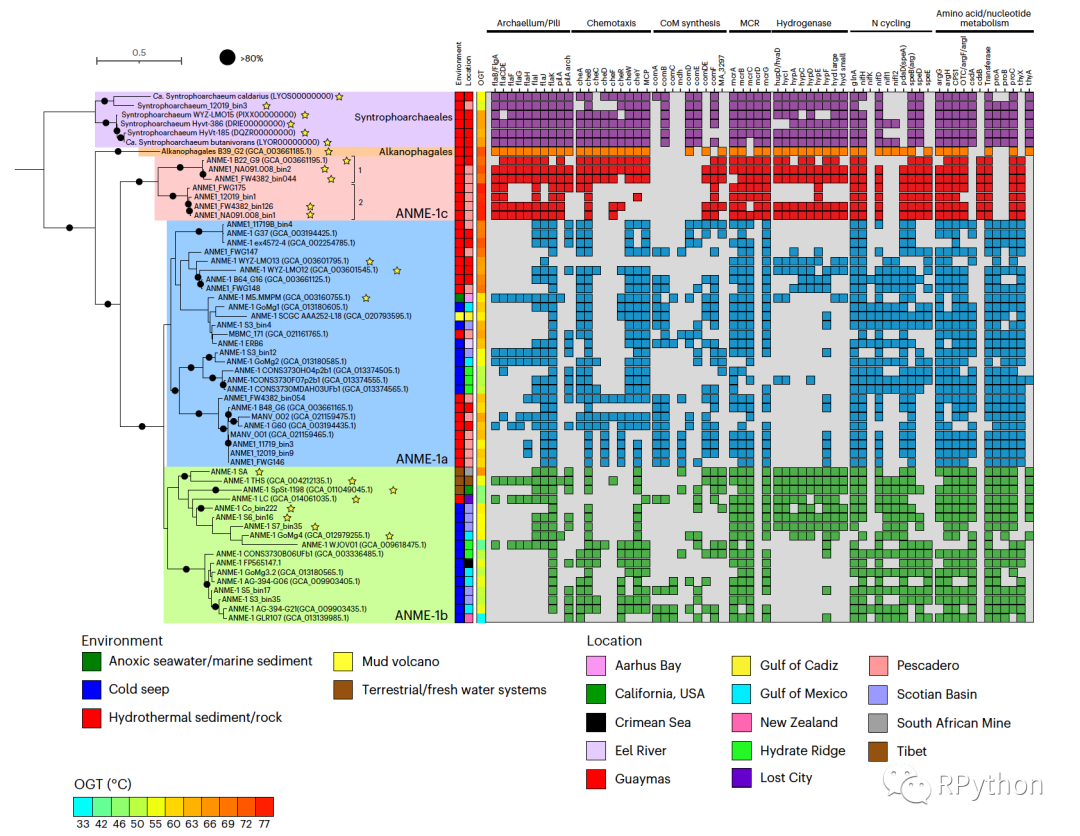
图片解读
- 首先,映入眼帘的就是一张绝美的进化树,一涉及到进化树,那么「Y叔」的
ggtree绝对是不逞多让,首屈一指的选择。 - 并且进化树里面基于不同的
节点node进行了进行了分组,添加了分组的信息,以及颜色的映射。 - 其次,进化树的右侧可以视为四个热图。
- 热图1和热图2,很明显都是一个分类变量(离散型变量)的热图。
- 热图3是一个数值型(连续型变量)的热图。
- 热图4是一个很大的分类变量(离散型变量)热图,并且基于前面的进化树的分类,进行了对应的颜色映射,同时热图的坐标轴进行了倒置,以及坐标轴进行了分类。
- 最后将各个部分的图片进行拼图。
本期推文的图片是基于ggplot2和ggtree实现的,虽然数据和配色略有不同,但是图片中的绝大部分的细节都实现了较高的还原。由于我并没有获取到作者文章的数据,因此我重新构建了包括:进化树,离散型变量的热图,连续型变量的热图的所有数据。
加载绘图所需的R包。
library(tidyverse)
library(ggtree)
library(treeio)
library(tidytree)
library(ggnewscale)
library(ggsci)
library(aplot)
library(patchwork)
library(ggh4x)构建测试数据。进化树的数据是我自己构建好的,我在树文件中添加了bootstrap信息,以便进行可视化。
set.seed(1115)
tree <- ggtree::read.tree(file = "example.tree")
as_tibble(tree) %>%
dplyr::mutate(bootstrap = sample(c(sample(1:30, 30, replace = T),
sample(31:75,60, replace = T),
sample(76:100, 35, replace = T)),
125, replace = F)) %>%
dplyr::mutate(star = ifelse(!is.na(label) &
str_split(label, pattern = "_", simplify = T)[,2] %>% as.numeric() > 4000,"star",NA))-> df_tip_data使用geom_hilight函数,基于节点进行添加颜色,使用geom_cladelab函数,基于节点信息添加文字描述信息。
ggtree(tree) %<+% df_tip_data +
# geom_tiplab(as_ylab = T, align = T) +
geom_tiplab(offset = 0.1) +
geom_tippoint() +
geom_nodepoint(aes(fill = cut(bootstrap, c(0, 30, 75, 100))),
shape = 21, size = 2) +
geom_hilight(node = 90, fill = "#e41a1c", color = NA, alpha = 0.3, to.bottom = T,extendto = 23.5) +
geom_hilight(node = 81, fill = "#377eb8", color = NA, alpha = 0.3, to.bottom = T, extendto = 23.5) +
geom_hilight(node = 66, fill = "#4daf4a", color = NA, alpha = 0.3, to.bottom = T, extendto = 23.5) +
geom_hilight(node = 112, fill = "#984ea3", color = NA, alpha = 0.3, to.bottom = T, extendto = 23.5) +
geom_hilight(node = 101, fill = "#ff7f00", color = NA, alpha = 0.3, to.bottom = T, extendto = 23.5) +
geom_cladelab(node = 90, label = "Group1", align = TRUE, vjust = 5, hjust = -1, barcolor = NA)+
geom_cladelab(node = 81, label = "Group2", align = TRUE, vjust = 5, hjust = -1, barcolor = NA)+
geom_cladelab(node = 66, label = "Group3", align = TRUE, vjust = 5, hjust = -1, barcolor = NA)+
geom_cladelab(node = 112, label = "Group4", align = TRUE, vjust = 5, hjust = -1, barcolor = NA)+
geom_cladelab(node = 101, label = "Group5", align = TRUE, vjust = 5, hjust = -1, barcolor = NA)+
geom_strip(taxa1 = "ASV_4428", taxa2 = "ASV_5", label = "1", offset = -2, offset.text = 0.5) +
geom_strip(taxa1 = "ASV_4011", taxa2 = "ASV_1359", label = "2", offset = -2, offset.text = 0.5) +
xlim(0,22)+
geom_treescale(x = 0) +
scale_fill_manual(values = c("white","grey","black"),
guide = "legend",
name = "Bootstrap Percentage(BP)",
breaks = c("(0,30]","(30,75]","(75,100]"),
labels = expression(BP*"<=30", 30<BP*"<=75", BP>75)) -> p1
p1则第一部分的进化树就绘制完成了, 如下所示:
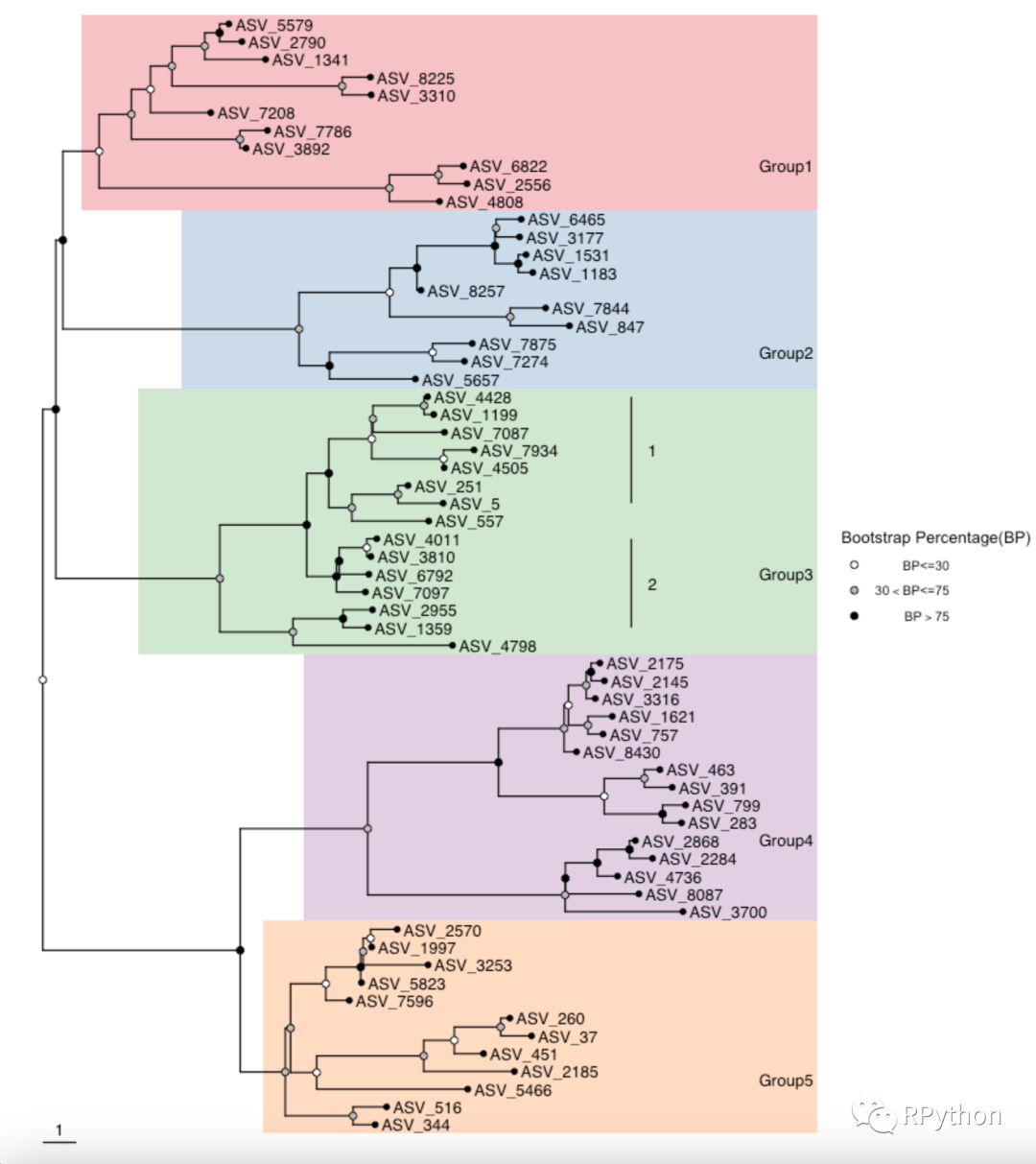
在绘制进化树的时候,往往会获取「进化树的label顺序」,Y叔早已考虑到了这个问题。 下面是基于「进化树的label顺序」再次构建热图数据,以及热图的可视化。 在这个热图数据的构建和绘制中,「在构建热图数据的技巧」,值得大家进行参考和借鉴。
# get tree order
get_taxa_name(p1) -> gene_name
# create heatmap data
heatmap_df <- data.frame(
gene_name = factor(gene_name, levels = rev(gene_name), ordered = T),
Environment = "Environment",
Variable1 = c(rep("A", 12), rep("B", 12), rep("C", 12), rep("D", 12), sample(LETTERS[1:5],size = 15, replace = T)),
Location = "Location",
Variable2 = c(rep("a", 10), rep("b", 10), rep("c", 10), rep("d", 10), sample(letters[1:5],size = 23, replace = T)),
none = "none",
Variable3 = NA,
OGT = "OGT",
Variable4 = seq(33, 100, length.out = 63)
)
# heatmap
ggplot(data = heatmap_df) +
geom_tile(aes(x = Environment, y = gene_name, fill = Variable1),
color = "black", alpha = 0.7, linewidth = 0.5) +
scale_fill_jco(name = "Environment") +
new_scale_fill() +
geom_tile(aes(x = Location, y = gene_name, fill = Variable2),
color = "black", alpha = 0.7, linewidth = 0.5) +
scale_fill_ucscgb(name = "Location")+
geom_tile(aes(x = none, y = gene_name),
fill = NA) +
new_scale_fill() +
geom_tile(aes(x = OGT, y = gene_name, fill = Variable4),
alpha = 0.85, color = "black") +
scale_fill_continuous(type = "viridis", name = "OGT") +
labs(x = "", y = "") +
scale_x_discrete(position = "top",
breaks = c("Environment", "Location", "OGT"),
labels = c("Environment", "Location", "OGT")) +
theme_bw() +
theme(
panel.border = element_blank(),
panel.grid = element_blank(),
axis.ticks = element_blank(),
axis.text.y = element_blank(),
axis.text.x = element_text(angle=90, hjust = 0)
) -> heatmap.p
heatmap.p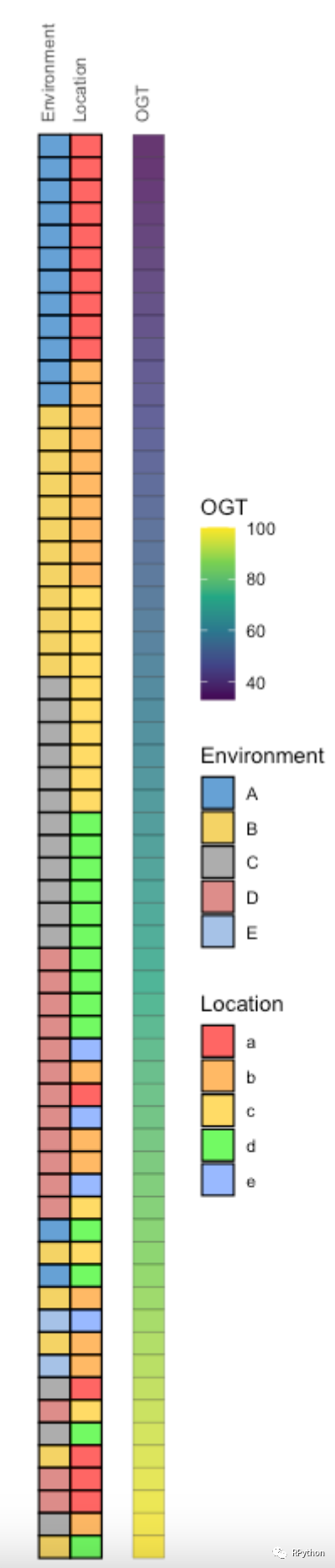
接下来才是重头戏,因为即将会有一个很大的热图出现。依旧是构建数据以及可视化。注意热图的坐标轴进行了倒置,并且对坐标轴进行了分类。
# heatmap
heatmap_df3 <- as.data.frame(
matrix(sample(c(NA, NA, "Yes"), size = 3780, replace = T),
nrow = 63, ncol = 60)
) %>%
set_names(paste(LETTERS[1:6],1:60,sep = "_")) %>%
dplyr::mutate(gene_name = factor(gene_name, levels = rev(gene_name), ordered = T)) %>%
dplyr::select(gene_name, everything())
heatmap_df4 <- heatmap_df3 %>%
tidyr::gather(key = "key", value = "value", -gene_name) %>%
tidyr::separate(col = key, into = c("Type"), sep = "_", remove = F) %>%
dplyr::filter(!is.na(value))
# Based on the node of the tree, the final heatmap data was constructed
groupClade(tree, .node = c(90, 81, 66, 112, 101)) %>%
as_tibble() %>%
dplyr::select(label, group) %>%
right_join(., heatmap_df4, by = c("label" = "gene_name")) %>%
dplyr::mutate(group = factor(group),
label = factor(label, levels = gene_name, ordered = T)) -> heatmap_df5
# heatmap
ggplot(data = heatmap_df5,
aes(interaction(key, Type), label)) +
geom_tile(aes(fill = group),
color = "black",alpha = 0.9, linewidth = 0.3) +
labs(x = "", y = "") +
guides(x = "axis_nested") +
scale_x_discrete(position = "top",
guide = guide_axis(position = "top")) +
scale_fill_manual(values = c(
"1" = "#e41a1c", "2" = "#377eb8", "3" = "#4daf4a", "4" = "#984ea3", "5" = "#ff7f00"
), name = "Group") +
theme_bw() +
theme(
axis.text.x.top = element_text(angle = 90),
panel.background = element_rect(fill = "#d9d9d9"),
panel.grid = element_blank(),
# legend.position = "none",
axis.text.y = element_blank(),
axis.ticks.y.left = element_blank(),
ggh4x.axis.nesttext.x = element_text(size = 10)
) -> heatmap.p2
heatmap.p2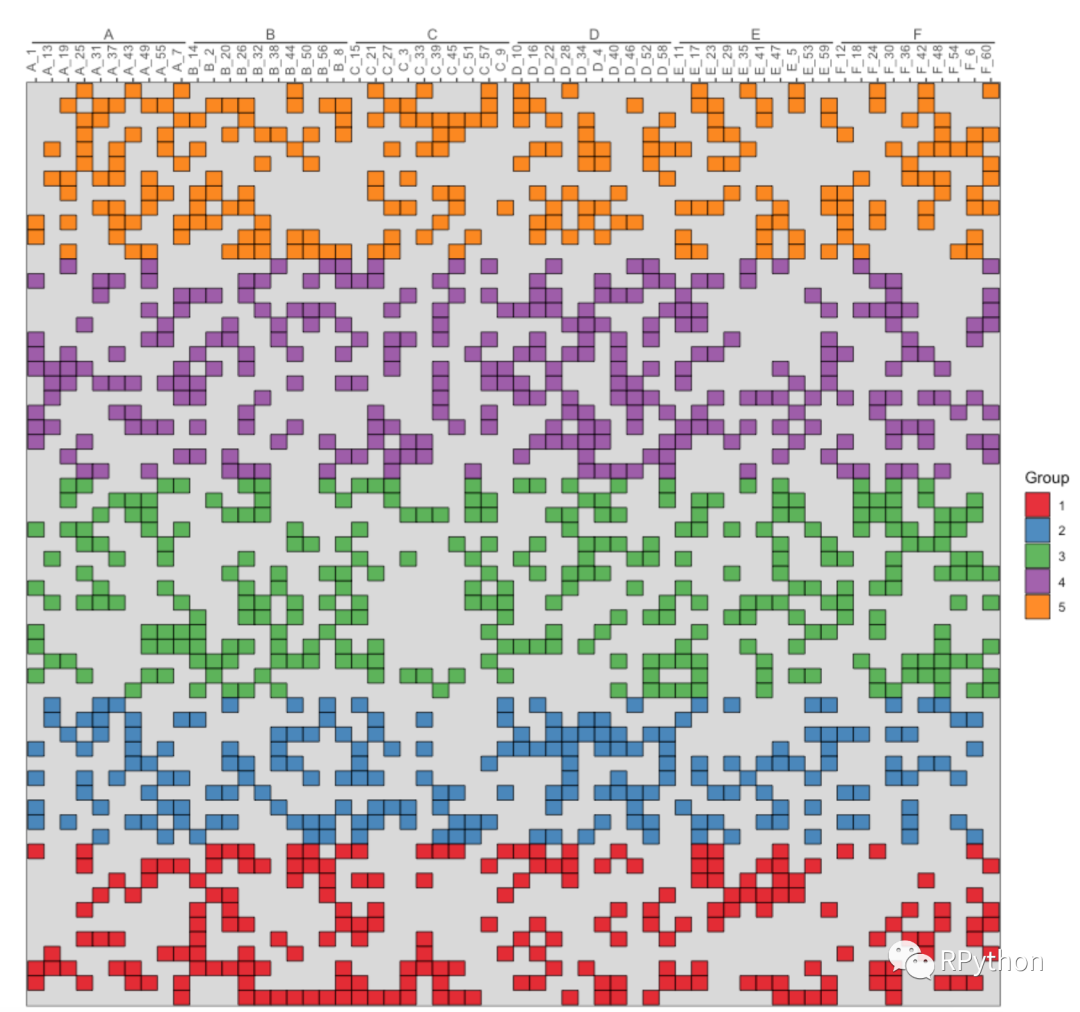
拼图以及保存结果
heatmap.p2 %>%
insert_left(., heatmap.p, width = 0.07) %>%
insert_left(., p1, width = 0.85)
ggsave(filename = "20230309_2.pdf",
height = 12.5,
width = 22)获取示例数据和绘图代码
# load R Package
library(tidyverse)
library(ggtree)
library(treeio)
library(tidytree)
library(ggnewscale)
library(ggsci)
library(aplot)
library(patchwork)
library(ggh4x)
set.seed(1115)
tree <- ggtree::read.tree(file = "example.tree")
as_tibble(tree) %>%
dplyr::mutate(bootstrap = sample(c(sample(1:30, 30, replace = T),
sample(31:75,60, replace = T),
sample(76:100, 35, replace = T)),
125, replace = F)) %>%
dplyr::mutate(star = ifelse(!is.na(label) &
str_split(label, pattern = "_", simplify = T)[,2] %>% as.numeric() > 4000,
"star",NA))-> df_tip_data
ggtree(tree) %<+% df_tip_data +
# geom_tiplab(as_ylab = T, align = T) +
geom_tiplab(offset = 0.1) +
geom_tippoint() +
geom_nodepoint(aes(fill = cut(bootstrap, c(0, 30, 75, 100))),
shape = 21, size = 2) +
geom_hilight(node = 90, fill = "#e41a1c", color = NA, alpha = 0.3, to.bottom = T,extendto = 23.5) +
geom_hilight(node = 81, fill = "#377eb8", color = NA, alpha = 0.3, to.bottom = T, extendto = 23.5) +
geom_hilight(node = 66, fill = "#4daf4a", color = NA, alpha = 0.3, to.bottom = T, extendto = 23.5) +
geom_hilight(node = 112, fill = "#984ea3", color = NA, alpha = 0.3, to.bottom = T, extendto = 23.5) +
geom_hilight(node = 101, fill = "#ff7f00", color = NA, alpha = 0.3, to.bottom = T, extendto = 23.5) +
geom_cladelab(node = 90, label = "Group1", align = TRUE, vjust = 5, hjust = -1, barcolor = NA)+
geom_cladelab(node = 81, label = "Group2", align = TRUE, vjust = 5, hjust = -1, barcolor = NA)+
geom_cladelab(node = 66, label = "Group3", align = TRUE, vjust = 5, hjust = -1, barcolor = NA)+
geom_cladelab(node = 112, label = "Group4", align = TRUE, vjust = 5, hjust = -1, barcolor = NA)+
geom_cladelab(node = 101, label = "Group5", align = TRUE, vjust = 5, hjust = -1, barcolor = NA)+
geom_strip(taxa1 = "ASV_4428", taxa2 = "ASV_5", label = "1", offset = -2, offset.text = 0.5) +
geom_strip(taxa1 = "ASV_4011", taxa2 = "ASV_1359", label = "2", offset = -2, offset.text = 0.5) +
xlim(0,22)+
geom_treescale(x = 0) +
scale_fill_manual(values = c("white","grey","black"),
guide = "legend",
name = "Bootstrap Percentage(BP)",
breaks = c("(0,30]","(30,75]","(75,100]"),
labels = expression(BP*"<=30", 30<BP*"<=75", BP>75)) -> p1
p1
# get tree order
get_taxa_name(p1) -> gene_name
# plot heatmap
heatmap_df <- data.frame(
gene_name = factor(gene_name, levels = rev(gene_name), ordered = T),
Environment = "Environment",
Variable1 = c(rep("A", 12), rep("B", 12), rep("C", 12), rep("D", 12), sample(LETTERS[1:5],size = 15, replace = T)),
Location = "Location",
Variable2 = c(rep("a", 10), rep("b", 10), rep("c", 10), rep("d", 10), sample(letters[1:5],size = 23, replace = T)),
none = "none",
Variable3 = NA,
OGT = "OGT",
Variable4 = seq(33, 100, length.out = 63)
)
ggplot(data = heatmap_df) +
geom_tile(aes(x = Environment, y = gene_name, fill = Variable1),
color = "black", alpha = 0.7, linewidth = 0.5) +
scale_fill_jco(name = "Environment") +
new_scale_fill() +
geom_tile(aes(x = Location, y = gene_name, fill = Variable2),
color = "black", alpha = 0.7, linewidth = 0.5) +
scale_fill_ucscgb(name = "Location")+
geom_tile(aes(x = none, y = gene_name),
fill = NA) +
new_scale_fill() +
geom_tile(aes(x = OGT, y = gene_name, fill = Variable4),
alpha = 0.85, color = "black") +
scale_fill_continuous(type = "viridis", name = "OGT") +
labs(x = "", y = "") +
scale_x_discrete(position = "top",
breaks = c("Environment", "Location", "OGT"),
labels = c("Environment", "Location", "OGT")) +
theme_bw() +
theme(
panel.border = element_blank(),
panel.grid = element_blank(),
axis.ticks = element_blank(),
axis.text.y = element_blank(),
axis.text.x = element_text(angle=90, hjust = 0)
) -> heatmap.p
insert_left(heatmap.p, p1, width = 6)
# heatmap2
heatmap_df3 <- as.data.frame(
matrix(sample(c(NA, NA, "Yes"), size = 3780, replace = T),
nrow = 63, ncol = 60)
) %>%
set_names(paste(LETTERS[1:6],1:60,sep = "_")) %>%
dplyr::mutate(gene_name = factor(gene_name, levels = rev(gene_name), ordered = T)) %>%
dplyr::select(gene_name, everything())
heatmap_df4 <- heatmap_df3 %>%
tidyr::gather(key = "key", value = "value", -gene_name) %>%
tidyr::separate(col = key, into = c("Type"), sep = "_", remove = F) %>%
dplyr::filter(!is.na(value))
# 基于进化树前面分组的数据
clade_1 <- tree_subset(tree, node = 90)
groupClade(tree, .node = c(90, 81, 66, 112, 101)) %>%
as_tibble() %>%
dplyr::select(label, group) %>%
right_join(., heatmap_df4, by = c("label" = "gene_name")) %>%
dplyr::mutate(group = factor(group),
label = factor(label, levels = gene_name, ordered = T)) -> heatmap_df5
glimpse(heatmap_df5)
ggplot(data = heatmap_df5,
aes(interaction(key, Type), label)) +
geom_tile(aes(fill = group),
color = "black",alpha = 0.9, linewidth = 0.3) +
labs(x = "", y = "") +
guides(x = "axis_nested") +
scale_x_discrete(position = "top",
guide = guide_axis(position = "top")) +
scale_fill_manual(values = c(
"1" = "#e41a1c", "2" = "#377eb8", "3" = "#4daf4a", "4" = "#984ea3", "5" = "#ff7f00"
), name = "Group") +
theme_bw() +
theme(
axis.text.x.top = element_text(angle = 90),
panel.background = element_rect(fill = "#d9d9d9"),
panel.grid = element_blank(),
# legend.position = "none",
axis.text.y = element_blank(),
axis.ticks.y.left = element_blank(),
ggh4x.axis.nesttext.x = element_text(size = 10)
) -> heatmap.p2
heatmap.p2 %>%
insert_left(., heatmap.p, width = 0.07) %>%
insert_left(., p1, width = 0.85)
ggsave(filename = "20230309_2.pdf",
height = 12.5,
width = 22)总结:这张Nature Microbiology的Figure 1,实际上是ggtree和ggplot2的综合使用。最为重要的是ggtree非常的好用,其次就是数据格式的问题。如果掌握了数据格式,那么复现文章的图片是非常容易的。
特别申明:本文为转载文章,转载自 RPython ,不代表贪吃的夜猫子立场,如若转载,请注明出处:https://mp.weixin.qq.com/s/A5vnKKXHE09LaQ8xwIBlhQ
 微信扫一扫
微信扫一扫  支付宝扫一扫
支付宝扫一扫